
Understanding ion diffusion is essential in battery materials research and characterizing diffusion pathways is critical for rational materials design and optimization. While using the diffraction technique for the characterization of battery materials, we could obtain the refined structure configuration via fitting the diffraction data, with the small-box or big-box approach, focusing on local structure or average structure. Accordingly, the ion diffusion behavior analysis could be conducted to yield the critical information about the ion diffusion pathway, based on experimental data, in contract to the similar information obtained via, e.g., molecular dynamics simulation.
Regarding the construction of the ion diffusion pathway from a structure configuration, a typical approach is based on the bond valence (BV) method. Details about the theory and formulation can be found in Refs. [1, 2]. In short, the backbone of the methodology can be summarized as follows,
-
The actual contribution to the valence of a central atom from a surrounding atom depends on the distance between them.
-
The overall valence of a central atom is the sum of contributions from all surrounding atoms, and in practice, a cutoff needs to be set to define the
surroundingatoms. -
Positions of ions are allowed to vary in reality and the diffusion pathway can be considered as the connection of those points yielding the same bond valence for ions.
-
Such an idea can be expanded to define an energy term that considers both the BV-determined energy and a Coulomb interaction term.
-
The ion diffusion pathway is then just the isosurface corresponding to a pre-defined energy value.
Such a theoretical framework based on the BV method for ion diffusion pathway construction is implemented in the BondStr program in the Fullprof Suite [3, 4]. Using the program is straightforward, but still there are some technical details to pay attention to get it working. First, we need to install the Fullprof Suite, which can be downloaded from their website [6]. The program is available on all Windows, Linux and MacOS – I am only using its Windows version and am not sure how robust it is working on Linux or MacOS. Once installed and launched, we should see the following interface of the program,

Regarding the purpose here, we want to click on the button as indicated by the red arrow shown in the picture to launch the Bond_Str GUI interface. This will bring up the following interface,

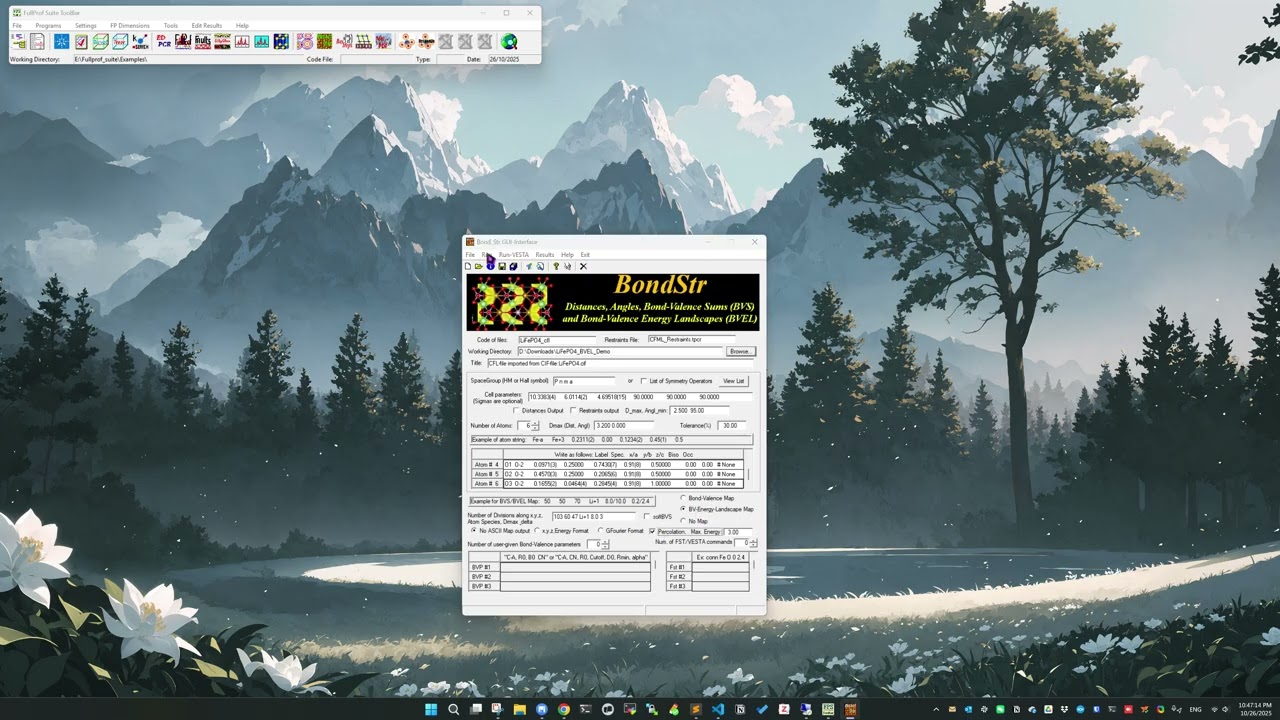
To use the program, there may be multiple workflow and here I am only putting down the one that I believe should be most common one. To start, we need a CIF file for the structure we want to analyze, and here I am using the example used in Ref. [4] and the CIF file can be downloaded here [5]. In the GUI, we go to File \(\Rightarrow\) Import CIF, and select the CIF we want to analyze. With the example CIF file here, the interface will now look like,

Looking at the atom lines in the red rectangle, we can see that the loaded atom lines are not following the expected format as given by the example atom string. Here we can manually edit those atom lines to make it look like the following,

where we change the Spec. part in each atom line to something like Li+1, FE+2, P+5 and O-2 to specify the ideal charge for each element type. Without any further changes, we can click on Run in the menu to start the bond valence calculation, in which case we will not calculate the isosurface regarding the ion diffusion pathway. Instead, the bond valence sum (BVS) for each atomic site (Wyckoff position) will be calculated and results can be found in the output .bvs file under the Working Directory (see the GUI for the specification of the Working Directory).
Here, it should be noticed that in the GUI, we have the
Code of files:entry which determines the stem name of output files. For example, if we follow the procedures above to calculate the BVS for the example CIF file provided, theCode of files:entry would beLiFePO4_cfland accordingly the BVS output file would beLiFePO4_cfl_sum.bvsunder theWorking Directory.
Next, to calculate the bond valence energy landscape (BVEL) map (see the #5 bullet point above), we want to put in some parameters in the Number of Division along x,y,z,... input box and click on the BV-Energy-Landscape Map option. The input box could be given a string like below,
103 60 47 Li+1 8.0 3
where the first three numbers specify the number of grid points along the x, y and z direction – in fact, I think it actually means the crystallographic a, b and c direction. The next part Li+1 specifies the atomic species to calculate and for Li-ion batteries, for sure the Li ions diffusion is what we care about. The next part 8.0 specifies the distance cutoff for considering surrounding atoms as neighbors. The last part 3 specifies the delta value for the energy such that the grid volume yielding an energy value smaller than Emin + delta will be considered as the mobile volume, i.e., the fractional volume that can be potentially considered as being able to contribute to the ion diffusion. Given a certain delta value, for sure, the bigger the fractional volume is, the better the expected ion diffusion will be.
The calculated fractional volume will be presented in the output window to be presented once the calculation is finished.
When the calculation is started, a terminal console will be brought up and once the calculation is finished, we can press the
[Enter]key to kill the terminal and the output window will be brought up automatically. To continue the program running, we need to close the output window.
The fraction volume mentioned here can be found in the output window following
Volume fraction for ion mobility in the unit cell:
Last, we may want to check Percolation box and input a value in the Max. Energy input box. The value here is also a delta value of energy. In this case, grid points yielding the energy lower than Emin + delta will be considered for establishing the percolation network of diffusion ions. For sure, a larger Max. Energy value would require longer computation time.
Here,
Eminmentioned in the context refers to the ground state energy.
A series of output will be generated along with the calculation and the most interesting output that we may want to check out is probably the .vesta file with which we can visualize the energy isosurface using VESTA. Opening the generated .vesta file in VESTA, we should be able to see the energy isosurface like shown below,

In fact, while loading the .vesta file into VESTA, by default we won’t see the isosurface as presented above. We need to make some changes to the isosurface parameters in VESTA. In the menu, we go to Objects \(\Rightarrow\) Properties \(\Rightarrow\) isosurfaces... to bring up the window as shown below,

Highlighting the part shown in the red rectangle, we can change the parameters for both the dropdown selection (Positive as shown in the picture) and the Isosurface level (-1, and the unit should be eV). The displayed isosurface is then the one corresponding to the percolation energy of -1 eV.
A demonstration video covering the basic procedures discussed in current post can be found below (click on the image to go to the YouTube video),
As a technical note, here I also want to put down the problem and solution that I recently found with the VESTA program. With the most recent Windows 11 system, VESTA would not launch normally – sometimes I had to launch the program multiple times before it can load in the CIF file or open the .vesta file without crashing. Not sure exactly from which version of Windows did the problem start to show up, but at least my current version of Windows 11 (Windows 11 Pro, Version 25H2, OS build 2600.6901) does show the issue with VESTA. In case the problem does happen, we can press [Start] to bring up the start menu, search for vesta and right click on the icon to further click on Open file location. Then we should see the VESTA shortcut in the appearing folder. Right click on the shortcut, click on Show more options and then click on Properties. Then go to the Compatibility tab and check Run this program in Compatibility mode for:. In the dropdown selection, we can choose Windows 8 (see the image below). This does solve the VESTA crashing issue for me.

Updates @ 11/03/2025 22:50:37 EST
The Bond_Str GUI sometimes would fail to handle CIF files. Even though, it is still hopeful that the backend executable can be used for the calculation. It is just that we need some more manual configurations. Initially, we can still try to import a CIF file following the instruction above. Ignoring whatever error message popping up, we can then go to the saving directory of the imported CIF file and there we will find a .cfl file with the same stem name as the import CIF file being generated in the same directory. Some of the contents in the generated .cfl file may be problematic blocking the BVS calculation. However, the atom lines can still be used. Next, we want to go back to the Bond_Str GUI, import a working CIF (e.g., the example CIF given above), follow the instructions above to make changes to atom labels, fill in necessary calculation parameters, perform the calculation, and then check out the accordingly generated .cfl file. Here, we want to save the working .cfl file to another file (to be used for our own sample configuration), go back to the previouly generated problematic .cfl file, copy the atom lines into the just saved new .cfl file to replace those atom lines originating from the working CIF. Then we also need to update the title, cell parameters, space group and those BVEL parameters to be consistent with our sample configuration. This way, we just manually make a working version of .cfl file which can be run with the bond_str executable from the command line. On Windows, if we right click on the FullProf_Suite shortcut from the start menu and trace all the way to the installation directory of the program by selecting Open file location, we should be able to find the bond_str executable. Not sure about MacOS or Linux but I guess it should not be super difficult to find the executable somewhere in the FullProf_Suite installation directory, e.g., on MacOS, if we choose to Show Contents by right clicking on the FullProf_Suite app inside the /Applications directory, we can launch the terminal app there in the directory and use the find command to look for bond_str, like find . -name 'bond_str'. Once we have the bond_str executable located, it can be executed from the terminal like,
/path/to/bond_str <name>.cfl
where <name>.cfl refers to the previously hand-crafted .cfl file. In fact, before running the program, for sure we need to manually add in charges of elements by editing the Species column in those atom lines (usually it is the 3rd column) to make it something like Li+1, Fe+2, O-2, etc.
Sometimes, we would still have issues with the calculation due to, e.g., potential parameters not defined in the database. For example, the Fe+2-N-3 pair potential is not defined, in which case we need to manually put in the parameters for those missing parameters for certain pairs. The potential parameter line should be like this,
BVELPAR FE+2 N-3 6 1.76 5 4.4621 1.96 1.59
The parameters come in the following order,
Cation Anion CN R0 Cutoff D0 Rmin Alpha
where the CN (for coordination number) and Cutoff parameters are not used for the potential calculation – they should still be provided for maintaining the format of the information provided in the output file [3]. The R0 parameter is just the R0 parameter in the BVS calculation [7]. The D0, Rmin and Alpha parameters are those in the Morse potential given in the following form,
N.B. We need to find the Morse potential parameters for a specific atom pair from the literature pool. Also, we need to pay attention to the unit of those parameters – bond_str is expecting D0 with the unit of eV, \(Å^{-1}\) for Alpha, and Å for Rmin. It should be noted that sometimes in the literature, the D0 parameter would be given in the unit of kcal/mol – refer to Ref. [8] for the conversion between energy units.
As the last step (hopefully), we also need to include potential parameters for those atom pairs of which the parameters already exist in the database (hard coded into the program) – the thing is, it seems that the program is expect the manual input for all pairs once the BVELPAR line is given in the .cfl file. Here comes a little trick – in the hand-crafted .cfl file, we can remove all those atoms involved in the pair for which the potential parameters are not defined, save the .cfl to a temporary version and run bond_str with the temporary .cfl file. Once the calculation is done, we will be brought up with an output window from which we can find the potential parameters for atom pairs, like shown below,
...
Bond-Valence Energy parameters (D0,Rmin,alpha) for Morse Potential: D0*[{exp(alpha(Rmin-d))-1}^2-1]
(data read from internal table, provided by the user or calculated from softBVS parameters)
Morse parameters obtained from internal table
Type 1: FE+2 with type 4: O-2
D0 = 1.69269 Rmin = 1.96005 Alpha = 2.08333
Av. Coord.= 5.74300 R0 = 1.57911 R-cutoff = 5.50000 => Reference: S. Adams and R. Prasada Rao, (2011) Phys. Status Solidi A 208, No. 8, 1746-1753
Cation (Eff. radius): FE+2( 1.260) Anion (Eff. radius): O-2 ( 1.330)
Type 2: LI+1 with type 4: O-2
D0 = 0.98816 Rmin = 1.94001 Alpha = 1.93798
Av. Coord.= 5.02100 R0 = 1.17096 R-cutoff = 5.50000 => Reference: S. Adams and R. Prasada Rao, (2011) Phys. Status Solidi A 208, No. 8, 1746-1753
Cation (Eff. radius): LI+1( 1.310) Anion (Eff. radius): O-2 ( 1.330)
Type 3: P+5 with type 4: O-2
D0 = 3.89635 Rmin = 1.44066 Alpha = 2.28833
Av. Coord.= 4.00000 R0 = 1.62038 R-cutoff = 5.00000 => Reference: S. Adams and R. Prasada Rao, (2011) Phys. Status Solidi A 208, No. 8, 1746-1753
Cation (Eff. radius): P+5 ( 1.100) Anion (Eff. radius): O-2 ( 1.330)
...
Grabbing the potential parameters from the output, we can then put in the BVELPAR line for all atom pairs. Then, bond_str should be able to run with the hand-crafted .cfl file.
References
[1] M. Sale and M. Avdeev, J. Appl. Cryst. (2012). 45, 1054–1056.
[2] S. Adams and R. P. Rao, Phys. Status Solidi A (2011) 208 (8), 1746–1753.
[5] https://code.ill.eu/scientific-software/crysfml/-/tree/Thierry/Program_Examples/BondStr/Examples
[6] https://www.ill.eu/sites/fullprof/
[7] https://www.iucr.org/resources/data/datasets/bond-valence-parameters
[8] https://wild.life.nctu.edu.tw/class/common/energy-unit-conv-table.html